
On the Case
By John A. Cieslak, MD, PhD; Elena G. Violari, MD; and Clifford Yang, MD
Radiology Today
Vol. 19 No. 5 P. 28
History
A 25-year-old man who is a nursing home resident with a past medical history of cerebral palsy presented to the emergency department with new-onset seizures. He was ventilator and enteral feed dependent and chronically nonverbal. A noncontrast CT head scan was obtained upon admission to evaluate for an intracranial hemorrhage.
Findings
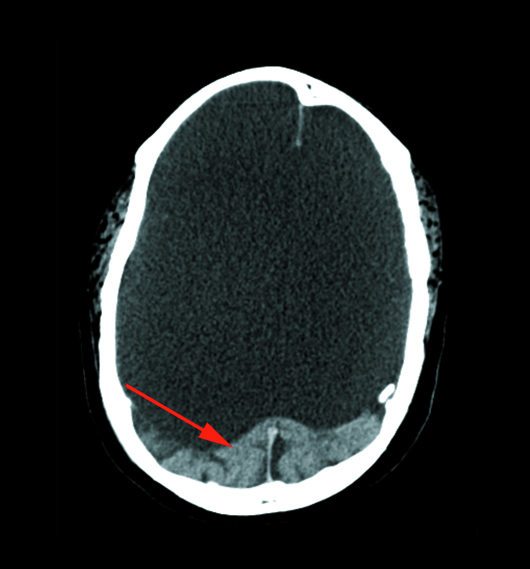
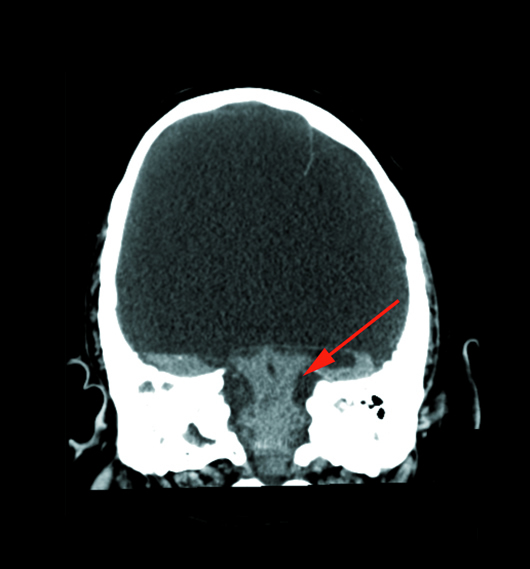
Helically acquired axial noncontrast CT brain image (Figure 1) demonstrated an absence of the cerebrum with a fluid-filled cranial vault. An occipital lobe remnant was seen (red arrow). Coronal CT brain image (Figure 2) through the level of the brainstem demonstrated a brainstem remnant (red arrow).
Figure 1 |
Figure 2 |
Diagnosis
Hydranencephaly (HE).
Discussion
HE is a rare cephalic disorder resulting from damage to or abnormal development of the fetal nervous system in utero or at birth, usually from a massive bilateral cerebral vascular infarct resulting in necrosis and absence of the cerebral hemispheres to varying degrees.1 The cerebral hemispheres are replaced with a cerebrospinal fluid-filled membranous sac that fills and sometimes expands the cranial vault. Typically, there is relative and variable preservation of the inferior frontal, temporal, and occipital lobes. The midbrain, cerebellum, thalami, basal ganglia, and choroid plexus are usually not involved.2,3 The incidence of HE is reported to be approximately one in 5,000.4 HE may affect only one hemisphere (hemihydrancencephaly), which is even rarer than the bilateral form, and typically allows for a better prognosis.5,6
Despite the growing number of case reports, most pathogenic, phenotypic, and prognostic aspects of HE remain controversial.1 Infection, such as toxoplasmosis, enterovirus, adenovirus, parvovirus, cytomegalovirus, herpes simplex virus, Epstein-Barr virus, and respiratory syncytial virus; radiation; fetal anoxia; medications or toxic exposures such as smoking, cocaine abuse, estrogens, and sodium valproate; twin-twin transfusion; cerebral hemorrhage; and genetic factors have all been proposed as initial causative factors. But the common finding observed on postnatal imaging studies is severe bilateral internal carotid artery (ICA) compromise with abnormalities ranging from ICA hypoplasia to aplasia.7-10
Two main hypotheses have been advanced to explain the severe brain impairment. One is the destructive theory, in which the HE occurs after the brain and ventricles have partially or fully formed and are then destroyed in utero, due to an encephaloclastic process.4 The second hypothesis refers to a dysontogenetic process with an early disruption of organogenesis.4,9
Diagnosis of HE can be made in utero by ultrasonography, though CT and MRI are also commonly used to make the diagnosis. Most of the case reports have been diagnosed between the 13th and the 26th week of pregnancy, during the second trimester of gestation.4 After delivery, the gold standard examinations are CT and MRI, which help differentiate HE from holoprosencephaly or severe congenital hydrocephalus.4
Prognosis for HE is poor; most affected children die before birth. Those who survive do not initially show evident neurological or clinical signs; archaic reflexes, leg, and arm movements are usually present at birth, as are sucking and swallowing reflexes.4 The signs become rapidly more pronounced after a few days, presenting with severe hypotonia, irritability, and seizures. In the children who survive, visual impairment, spastic diplegia, and cognitive delay are expected.
— John A. Cieslak, MD, PhD, is a diagnostic radiology resident at Northwestern University.
— Elena G. Violari, MD, is a radiology resident at the University of Connecticut.
— Clifford Yang, MD, is an associate professor of radiology at the University of Connecticut.
References
1. Cecchetto G, Milanese L, Giordano R, Viero A, Suma V, Manara R. Looking at the missing brain: hydranencephaly case series and literature review. Pediatr Neurol. 2013;48(2):152-158.
2. Hamby WB, Krauss RF, Beswick WF. Hydranencephaly; clinical diagnosis; presentation of 7 cases. Pediatrics. 1950;6(3):371-383.
3. Lam YH, Tang MH. Serial sonographic features of a fetus with hydranencephaly from 11 weeks to term. Ultrasound Obstet Gynecol. 2000;16(1):77-79.
4. Pavone P, Praticò AD, Vitaliti G, et al. Hydranencephaly: cerebral spinal fluid instead of cerebral mantles. Ital J Pediatr. 2014;40:79.
5. Greco F, Finocchiaro M, Pavone P, Trifiletti RR, Parano E. Hemihydranencephaly: case report and literature review. J Child Neurol. 2001;16(3):218-221.
6. Pavone P, Nigro F, Falsaperla R, et al. Hemihydranencephaly: living with half brain dysfunction. Ital J Pediatr. 2013;39:3.
7. Jordan L, Raymond G, Lin D, Gailloud P. CT angiography in a newborn child with hydranencephaly. J Perinatol. 2004;24(9):565-567.
8. McAbee GN, Chan A, Erde EL. Prolonged survival with hydranencephaly: report of two patients and literature review. Pediatr Neurol. 2000;23(1):80-84.
9. Quek YW, Su PH, Tsao TF, et al. Hydranencephaly associated with interruption of bilateral internal carotid arteries. Pediatr Neonatol. 2008;49(2):43-47.
10. Sepulveda W, Cortes-Yepes H, Wong AE, Dezerega V, Corral E, Malinger G. Prenatal sonography in hydranencephaly: findings during the early stages of disease. J Ultrasound Med. 2012;31(5):799-804.
