By Mathieu Sabbagh, DO; Hadi Bazzi, DO; and Brett Arnkoff, MD
Radiology Today
Vol. 18 No. 8 P. 28
History
A 21-year-old woman presented for an outpatient nonenhanced CT of the abdomen and pelvis due to left flank pain. The imaging study was followed by a contrast-enhanced MRI of the abdomen. Laboratory studies, including a comprehensive metabolic panel, complete blood count, and 24-hour urine metanephrines and vanillylmandelic acid, were within normal limits.
Findings
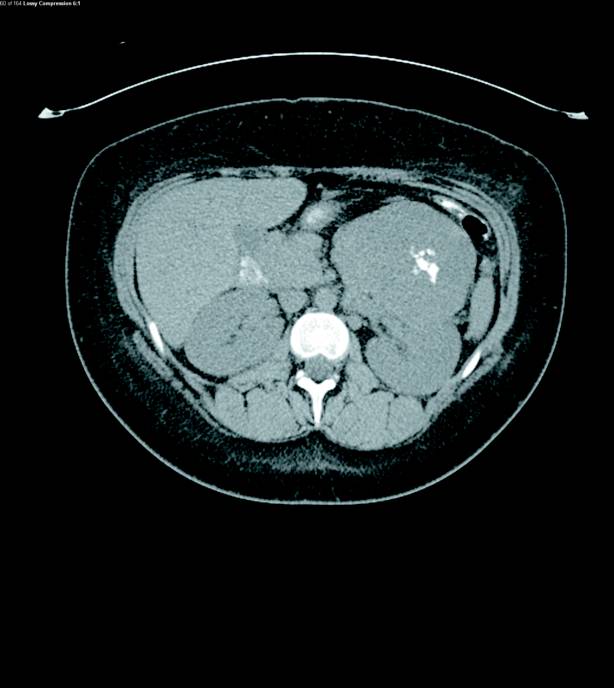
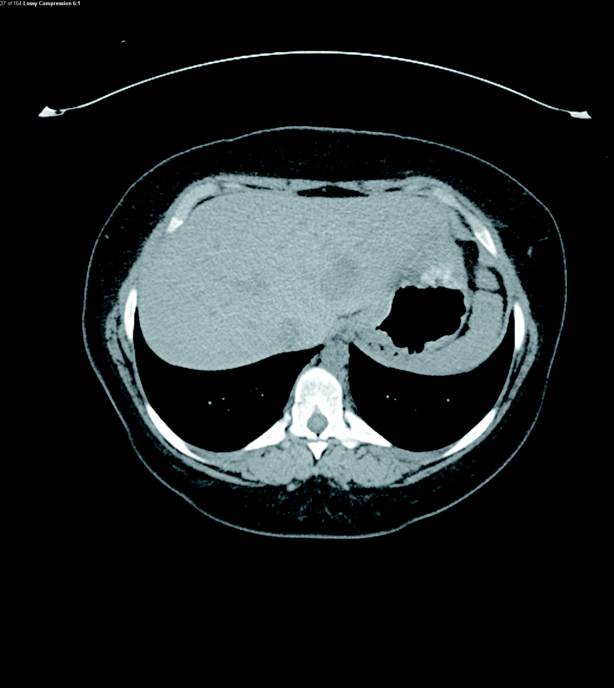
Nonenhanced CT abdomen and pelvis demonstrated a large heterogeneous mass arising from the left adrenal gland measuring up to 9.3 cm in diameter. Coarse intratumoral calcifications were present (Figure 1). At least four liver hypoattenuating lesions were seen in segments 2, 3, 5, and 6. The largest lesion was found in segment 2 of the left lobe of the liver measuring up to 3.5 cm in diameter (Figure 2).
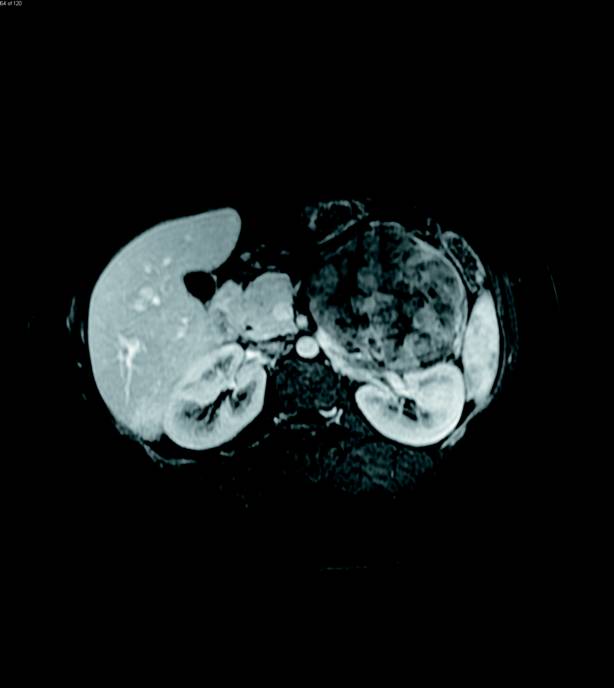
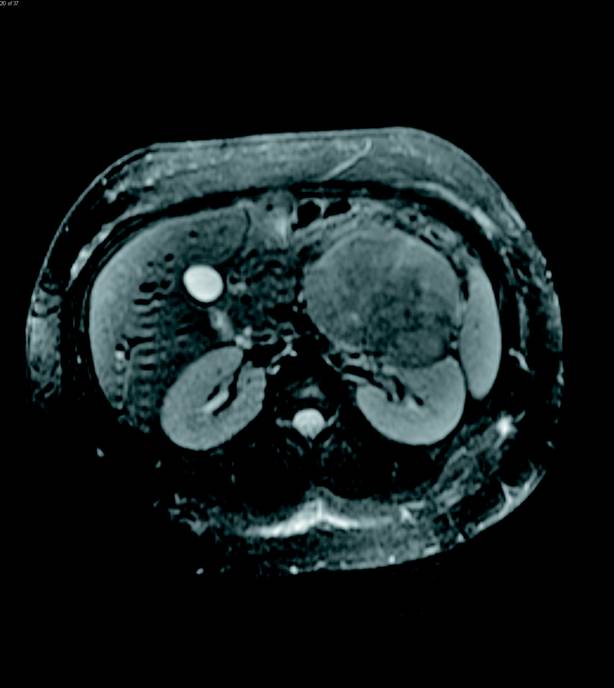
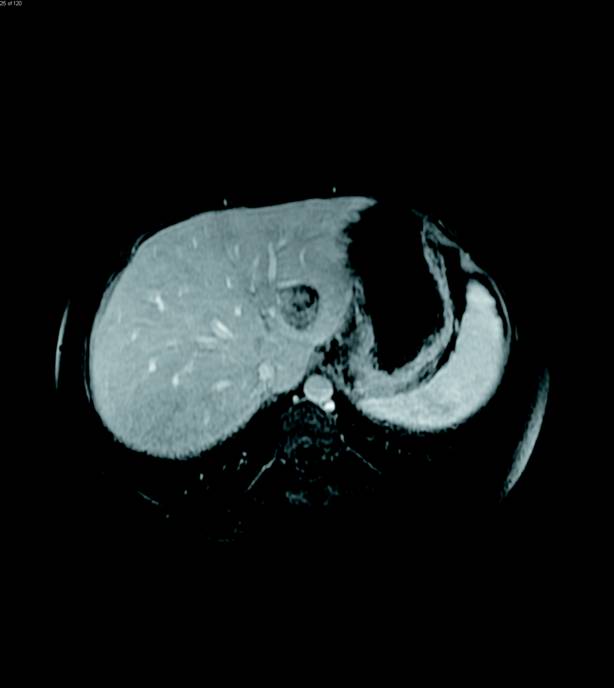
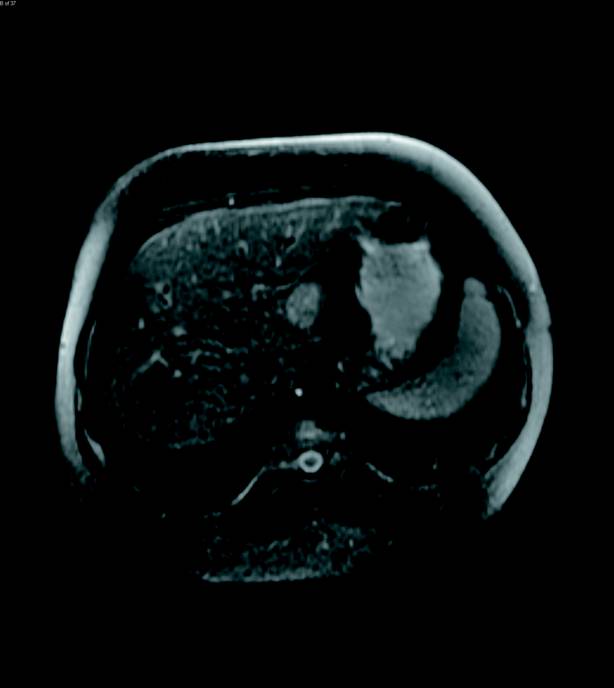
Contrast-enhanced MRI of the abdomen was then performed and illustrated a large, solid heterogeneously enhancing mass originating from the left adrenal gland with only slight T2 hyperintensity (Figures 3 and 4). The enhancement progressed on delayed imaging with central necrosis noted. Several enhancing, T2 hyperintense lesions in the liver were also identified, demonstrating T2 signal isointense to the spleen (Figures 5 and 6).
Diagnosis
Left adrenal cortical carcinoma (ACC) with metastases to the liver.
Discussion
ACC is a rare tumor of the adrenal cortex that affects one to two persons per million per year with a slight predominance in women. ACC accounts for only 0.02% of malignant tumors with approximately 70% of adrenal masses being benign.1 There are two peaks of incidence: the first and fourth decades of life.2,3 The classic clinical presentation is a palpable abdominal mass with abdominal or flank pain. Today, ACCs are often found incidentally on imaging studies.2
ACC may be related to multiple hereditary syndromes including Beckwith-Wiedemann, Carney complex, familial adenomatous polyposis, Li-Fraumeni, and multiple endocrine neoplasia Type 1.2
ACC can secrete a single hormone or a combination of hormones including androgens, aldosterone, cortisol, and estrogen. Patients may present with Cushing syndrome, Conn syndrome, hypertension, or virilization.1,2 Metastatic disease may be present at the time of presentation. ACC may metastasize to liver, lung, bone, and adjacent lymph nodes. The most common site of metastasis is the liver, and hepatic metastases are vascular with early arterial enhancement on contrast-enhanced studies.2,3 ACC has a known potential of venous invasion with tumor thrombus extension into the inferior vena cava.3
Imaging features of ACC include irregular margins, displacement of adjacent structures, heterogeneous enhancement, classic internal calcifications, venous extension, central necrosis, hemorrhage, and invasion of neighboring tissue.2 Most ACCs are larger than 4 cm when first discovered.3
ACC will demonstrate heterogeneous enhancement and appear T1 isointense or hypointense, although it may be hyperintense on T1 weighted images from hemorrhage. T2 weighted images will show heterogeneous hyperintense signal, although less T2 hyperintense than would be expected with a pheochromocytoma.
Differential diagnosis includes adrenal adenoma, pheochromocytoma, and adrenal metastasis. Adenomas are usually smaller than 4 cm and may have MR chemical shift artifact or measure 10 or less Hounsfield Units on nonenhanced CT if lipid rich.1,2 Pheochromocytomas classically are markedly T2 hyperintense, with most being functional and secreting catecholamines. Patients will present with new onset paroxysmal hypertension.2,3 Metastatic lesions are often bilateral and will be associated with a known primary tumor.2
Treatment for ACC is surgical excision. Chemotherapy with the adrenolytic agent Mitotane may be used for symptomatic management and improved survival for both metastatic and recurrent disease. Radiation therapy is reserved for patients with high risk of recurrence or treatment of bone metastasis.2
— Mathieu Sabbagh, DO, is an R2 diagnostic radiology resident at Michigan State University — Garden City Hospital Division.
— Hadi Bazzi, DO, is an R4 diagnostic radiology resident at Michigan State University — Garden City Hospital Division.
— Brett Arnkoff, MD, is a clinical assistant professor at Michigan State University.
References
1. Szolar DH, Korobkin M, Reittner P, et al. Adrenocortical carcinomas and adrenal pheochromocytomas: mass and enhancement loss evaluation at delayed contrast-enhanced CT. Radiology. 2005;234(2):479-485.
2. Bharwani N, Rockall AG, Sahdev A, et al. Adrenocortical carcinoma: the range of appearances on CT and MRI. AJR Am J Roentgenol. 2011;196(6):W706-W714.
3. Johnson PT, Horton KM, Fishman EK. Adrenal mass imaging with multidetector CT: pathologic conditions, pearls, and pitfalls. Radiographics. 2009;29(5):1333-1351.