By Gaurish G. Surlakar, MD
Radiology Today
Vol. 19 No. 11 P. 30
History
A 57-year-old man presented for imaging with a past medical history of a seizure disorder since childhood, following an episode of meningitis at 5 years of age. Prior imaging studies were not available. The patient underwent an MRI and MR angiogram (MRA) of the brain.
Findings
Fluid-attenuated inversion recovery (FLAIR) and T2 sequences demonstrated encephalomalacia involving the left frontal, temporal, and anterior parietal cortex with ex vacuo dilatation of the left lateral ventricle (Figures 1 and 2). Left-sided atrophy of the caudate, thalamus, and basal ganglia was also noted. Corpus callosal thinning was present as well as left-sided midbrain and pontine atrophy (Figure 3). Crossed cerebellar atrophy was noted with atrophy of the superior cerebellar peduncle and prominence of the cerebellar folia in the right cerebellar hemisphere causing mild asymmetric dilatation of the fourth ventricle (Figure 4). MRA revealed that the intracranial left internal carotid artery was irregularly narrowed (Figure 5). Left middle cerebral artery and anterior cerebral artery demonstrated significant reduction in the luminal calibers and irregular patchy flow signals on time-of-flight angiography (Figure 6).
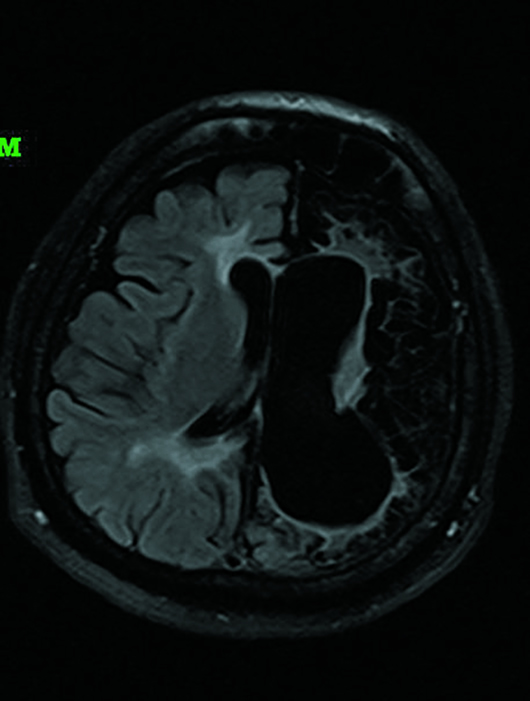
Figure 1
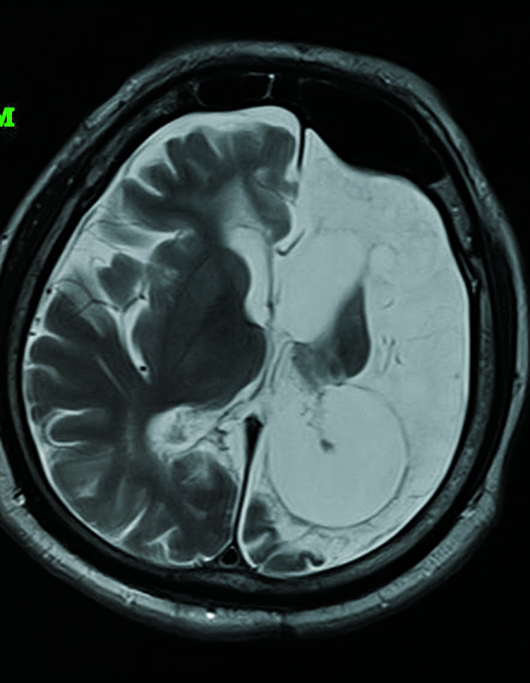
Figure 2
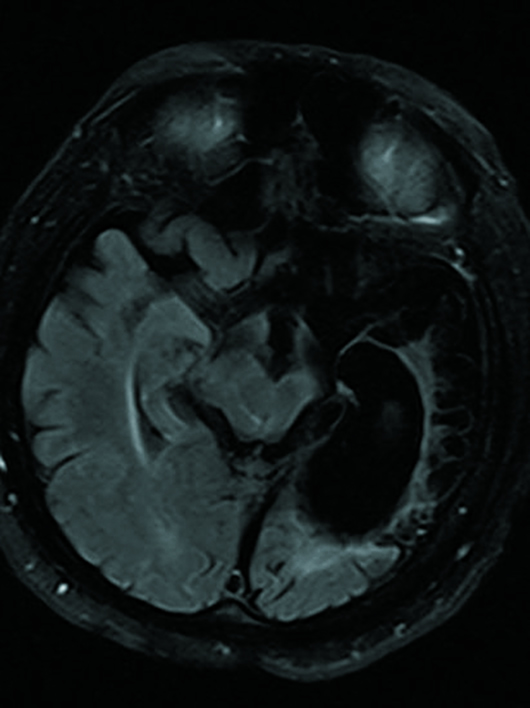
Figure 3
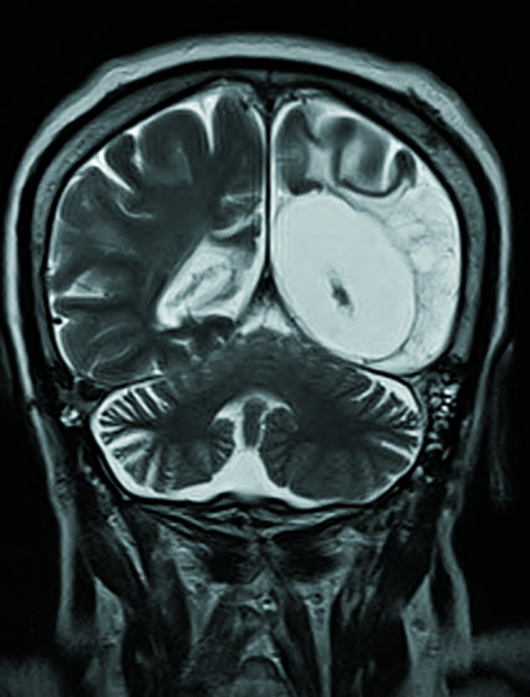
Figure 4
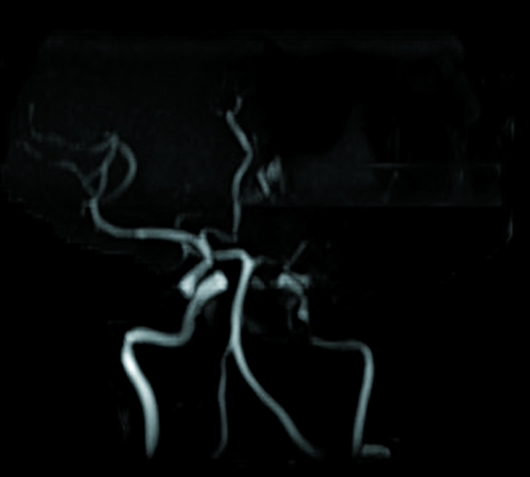
Figure 5
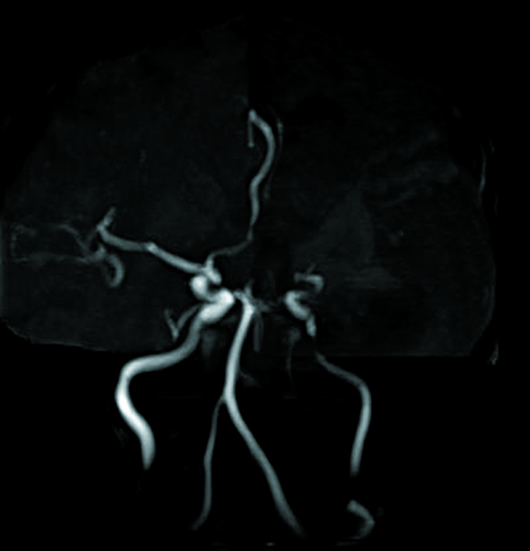
Figure 6
Diagnosis
Rasmussen’s encephalitis (RE) with long-term sequalae.
Discussion
RE is a rare inflammatory neurological disease that occurs mainly in children, characterized by frequent and severe seizures, loss of motor skills and speech, hemiparesis, encephalitis, and dementia. The mean age of presentation is between 6 and 8 years, with both sexes equally affected. However, detection of RE in adults is increasing with routine MRI investigations for intractable seizure.
Etiology of RE is unknown, with earlier studies suggesting the role of viral infections, while others describing it as an autoimmune phenomenon involving antibodies against a protein of the glutamate receptor. According to a recent concept, cytotoxic T cell reaction against the neuron leads to expression of major histocompatibility complex class 1 and apoptotic neuronal death, resulting in progressive deterioration of neurological status.
Clinically, RE presents as epilepsia partialis continua or generalized status epilepticus followed by hemiparesis and cognitive impairment, which gradually progresses over time. Seizures at presentation can be intractable, despite aggressive medical management. Apart from seizures, patients may have hemiparesis and speech disturbances, as well as hemianopia—secondary to unilateral cerebral involvement. Mental deterioration over time may also be seen, especially in patients presenting later in adolescence.
Diagnosis of RE is established based on characteristic clinical, radiological, and pathological features with more emphasis on clinicoradiological features; a brain biopsy, due to its invasive nature, is not done in all the cases. Imaging also plays a pivotal role by excluding other causes and is used to monitor disease progress.
RE imaging features are usually isolated to a single hemisphere; however, bilateral RE has also been described.
In pediatric unihemispheric progressive cerebral atrophy, RE should be considered in the differential diagnosis, especially if there is no intracranial calcification or contrast enhancement.
CT imaging may not show any specific features in early-stage RE; however, patchy hypodense attenuation areas similar to viral encephalitis may be seen. Late-stage disease may show unilateral cortical atrophy. CT perfusion may show decreased cerebral blood flow.
MRI abnormalities of RE evolve from the initial unihemispheric swelling (high signal on T2/FLAIR) and progress to severe cortical atrophy (disappearance of abnormal signal). In later disease, MRI also demonstrates ex-vacuo ventricular dilatation and T2 hyperintense signal areas in the affected hemisphere. Restricted diffusion may be seen in altered signal areas but is usually not a feature in chronic disease with cerebral hemiatrophy. Gadolinium-enhanced imaging has not been found to have any added advantage for establishing the diagnosis. Rare reports suggest instances of gadolinium-associated exacerbation of seizure frequency and neurologic deficits.
MRA is useful in excluding large to medium vessel vasculitis as the cause of signal changes, including extremely rare unilateral Moyamoya disease.
Differential diagnoses for RE include Dyke-Davidoff-Masson syndrome, Sturge-Weber syndrome, hemimegalencephaly, and unihemispheric cerebral vasculitis.
Outcome of RE is disappointing, though early initiation of therapy can delay the progression of disease. Treatment includes immunosuppressive and immunomodulator regimens in the form of steroids, immunoglobulins, plasma exchange, and tacrolimus.
— Gaurish G. Surlakar, MD, works at Radiance Diagnostics in Goa, India.
Resources
1. Rasmussen T, Olszewski J, Lloyd-Smith D. Focal seizures due to chronic localized encephalitis. Neurology. 1958;8(6):435-445.
2. Bien CG, Granata T, Antozzi C, et al. Pathogenesis, diagnosis and treatment of Rasmussen encephalitis: a European consensus statement. Brain. 2005;128(Pt 3):454-471.
3. Hunter GR, Donat J, Pryse-Phillips W, Harder S, Robinson CA. Rasmussen’s encephalitis in a 58 year old female: still a variant? Can J Neurol Sci. 2006;33(3):302-305.
4. Hart YM, Andermann F, Fish DR, et al. Chronic encephalitis and epilepsy in adults and adolescents: a variant of Rasmussen’s syndrome? Neurology. 1997;48(2):418-424.