By Midhir Patel, MD; Hieu Diep, MD; and Do Hyun Kim
Radiology Today
Vol. 22 No. 8 P. 34
History
A 40-year-old short-stature male with a past medical history of acute myelogenous leukemia (AML) with monosomy 7/del (7q) with TP53 mutation and pancreatic insufficiency presented to the ED with a three-week history of fever. Labs revealed pancytopenia with an absolute neutrophil count of 0.02 X 109/L. A bone marrow biopsy showed hypocellularity. CT of the abdomen was obtained. Patient was admitted for neutropenic fever.
Findings
Axial, sagittal, and coronal noncontrast CT images of the abdomen demonstrate homogenous diffuse fatty replacement and enlargement of the pancreas (Figures 1-3). Additional high-resolution CT image of the chest demonstrates normal lung parenchyma without bronchiectasis or bronchial wall thickening (Figure 4).
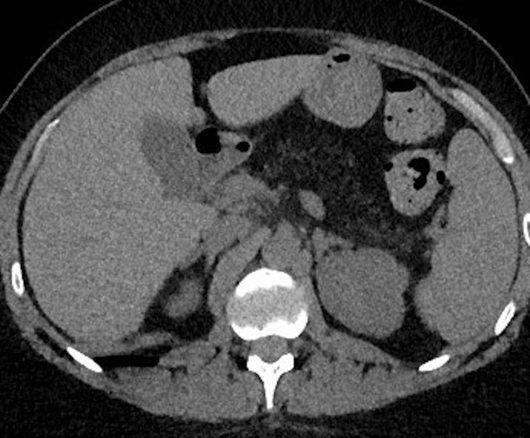
Figure 1
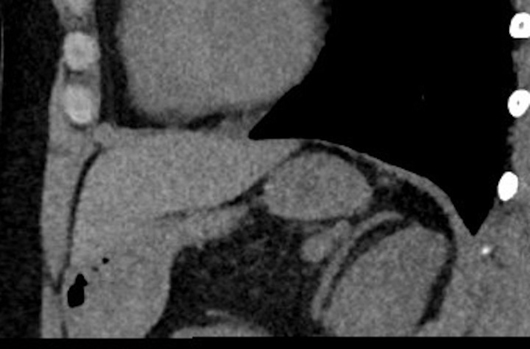
Figure 2
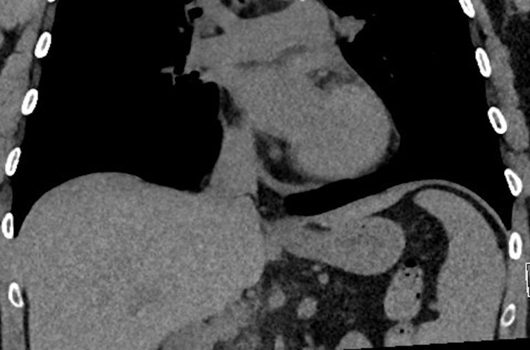
Figure 3
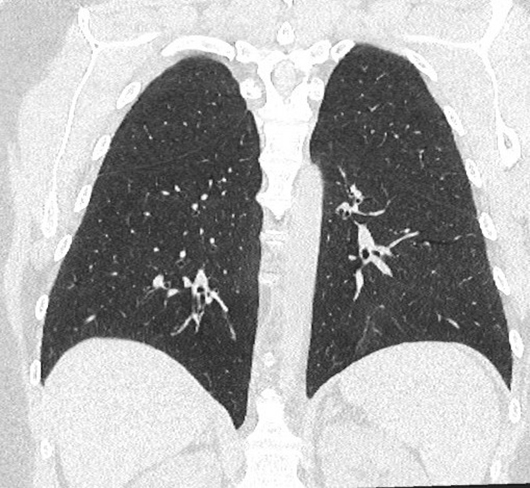
Figure 4
Diagnosis
Shwachman-Diamond syndrome (SDS). Genetic testing confirmed.
Discussion
SDS is a rare autosomal recessive disorder with an estimated incidence of 1 in 77,000 newborns in North America, although this number is suspected to be an underestimate since the signs and symptoms of SDS are variable and mild in some individuals. While the disorder is not predisposed to any specific racial population, a male-to-female ratio of 1.7:1 has been reported. SDS classically presents in early childhood; some studies have shown a median age of diagnosis as late as 11.1 years.
In approximately 80% of cases, individuals inherit two pathogenic variants of the SBDS gene on chromosome 7q11. This gene is responsible for facilitating the assembly between the 60S and 80S ribosomal subunits, as well as stabilizing the mitotic spindle. Thus, mutations in this gene lead to dysfunctional ribosome biogenesis and mitosis, which contribute to the loss of function of hematopoietic stem cells, the pancreatic exocrine system, and other important systems involved with cell division and growth. Less common mutations in other genes, such as DNAJC21 and EFL1, are also associated with ribosome assembly or protein translation. Some rare cases of SDS have been found to occur in an autosomal dominant fashion, but these typically result from de novo mutations in individuals with no previous family history of SDS.
Up to 81% of affected individuals have been shown to present with neutropenia with a high risk of life-threatening infections. Aplasia of the pancreatic acinar cells and diffuse fatty infiltration appear to play a role in the development of pancreatic dysfunction, leading to steatorrhea in a majority of patients. Macrocytic anemia, thrombocytopenia, diarrhea, and short stature are common manifestations as well. Affected individuals are at higher risk of malignant transformation, with approximately 1% higher risk per year for developing AML or myelodysplastic syndromes. Presentation can occur at any time of life, but it most commonly occurs during childhood.
Clinical signs are important in making a diagnosis of SDS. However, variability in clinical presentation makes a clinical diagnosis challenging, as only up to 51% of patients have been shown to exhibit the “classic” presentation of both neutropenia and steatorrhea. Consequently, SDS is arguably best confirmed by genetic testing for the SBDS gene mutation, while clinical signs and imaging offer a strong preliminary diagnosis. Currently, the primary mechanism of suspecting SDS involves imaging of the pancreas, which can support clinical diagnosis of SDS. However, imaging findings are not specific since conditions such as cystic fibrosis and Pearson syndrome can have similar findings.
Pancreatic ultrasonography in patients after the early infantile period has shown adequate sensitivity in detecting pathologic changes of SDS, which primarily includes increased pancreatic echogenicity consistent with pancreatic lipomatosis and depletion of acini. On the other hand, CT of the abdomen demonstrates homogenous fatty replacement and enlargement of the pancreas. These findings can overlap with cystic fibrosis (pancreatic atrophy, diffuse lipomatosis, and/or cystosis) and Pearson syndrome (fibrosis without lipomatosis). Differentiating unique features of cystic fibrosis are chronic lung infections that lead to pulmonary findings on imaging such as bronchiectasis, air trapping, and bronchial wall thickening; these are absent in SDS.
Current treatment options for SDS include pancreatic enzyme replacement therapy, packed red blood cell transfusions, and granulocyte colony-stimulating factor, which are targeted toward resolving the symptoms rather than the underlying disorder. Hematopoietic cell transplantation can potentially cure the hematologic abnormalities of SDS but does not treat pancreatic exocrine dysfunction or phenotypic changes commonly seen in SDS. Because diagnosing mild cases of SDS is difficult, it is challenging to determine the actual prognosis of SDS, since more severe cases tend to be more accurately diagnosed and can skew results. The most common causes of death in individuals with SDS are infections and hemorrhage. Although the prognosis is not clear, with one study estimating the life expectancy to be about 35 years, many individuals with SDS can live normal lives with routine monitoring.
— Midhir Patel, MD, is a radiologist at AdventHealth Orlando.
— Hieu Diep, MD, is a radiology resident at AdventHealth Orlando.
— Do Hyun Kim is a third-year medical student at Loma Linda University School of Medicine in California.
Resources
1. Boocock GR, Morrison JA, Popovic M, et al. Mutations in SBDS are associated with Shwachman-Diamond syndrome. Nat Genet. 2003;33(1):97-101.
2. Dale DC, Bolyard AA, Schwinzer BG, et al. The Severe Chronic Neutropenia International Registry: 10-year follow-up report. Support Cancer Ther. 2006;3(4):220-231.
3. Myers KC, Bolyard AA, Otto B, et al. Variable clinical presentation of Shwachman-Diamond syndrome: update from the North American Shwachman-Diamond Syndrome Registry. J Pediatr. 2014;164(4):866-870.
4. Adachi M, Tachibana K, Asakura Y, Aida N. Usefulness of pancreatic ultrasonography in the diagnosis of Shwachman-Bodian-Diamond syndrome. Acta Paediatr. 2005;94(11):1686-1690.
5. Hall GW, Dale P, Dodge JA. Shwachman-Diamond syndrome: UK perspective. Arch Dis Child. 2006;91(6):521-524.